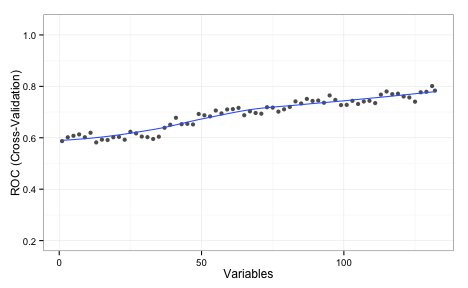
The values shown on the plot are the average area under the ROC curve measured using 5 repeats of 10-fold cross-validation. This implies that the full set of predictors is required. I should also note that, even with the entire predictor set, the KNN model did worse than LDA and random forests.
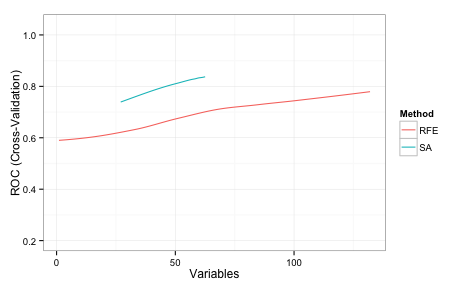
While RFE couldn't find a better subset of predictors, that does not mean that one doesn't exist. Can simulated annealing do better?
The code to load and split the data are in the AppliedPredictiveModeling package and you can find the markdown for this blog post linked at the bottom of this post. We have a data frame called training that has all the data used to fit the models. The outcome is a factor called Class and the predictor names are in a character vector called predVars. First, let's define the resampling folds.
Let's define the control object that will specify many of the important options for the search. There are a lot of details to spell out, such as how to train the model, how new samples are predicted etc. There is a pre-made list called caretSA that contains a starting template.
[1] "fit" "pred" "fitness_intern" "fitness_extern"
[5] "initial" "perturb" "prob" "selectIter"
These functions are detailed on the packages web page. We will make a copy of this and change the method of measuring performance using hold-out samples:
knnSA <- caretSA
knnSA$fitness_extern <- twoClassSummary
This will compute the area under the ROC curve and the sensitivity and specificity using the default 50% probability cutoff. These functions will be passed into the safs function (see below)
safs will conduct the SA search inside a resampling wrapper as defined by the index object that we created above (50 total times). For each subset, it computed and out-of-sample (i.e. "external") performance estimate to make sure that we are not overfitting to the feature set. This code should reproduce the same folds as those used in the book:
library(caret)
set.seed(104)
index <- createMultiFolds(training$Class, times = 5)
Here is the control object that we will use:
ctrl <- safsControl(functions = knnSA,
method = "repeatedcv",
repeats = 5,
## Here are the exact folds to used:
index = index,
## What should we optimize?
metric = c(internal = "ROC",
external = "ROC"),
maximize = c(internal = TRUE,
external = TRUE),
improve = 25,
allowParallel = TRUE)
The option improve specifies how many SA iterations can occur without improvement. Here, the search restarts at the last known improvement after 25 iterations where the internal fitness value has not improved. The allowParallel options tells the package that it can parallelize the external resampling loops (if parallel processing is available).
The metric argument above is a little different than it would for train or rfe. SA needs an internal estimate of performance to help guide the search. To that end, we will also use cross-validation to tune the KNN model and measure performance. Normally, we would use the train function to do this. For example, using the full set of predictors, this could might work to tune the model:
train(x = training[, predVars],
y = training$Class,
method = "knn",
metric = "ROC",
tuneLength = 20,
preProc = c("center", "scale"),
trControl = trainControl(method = "repeatedcv",
## Produce class prob predictions
classProbs = TRUE,
summaryFunction = twoClassSummary,))
The beauty of using the caretSA functions is that the ... are available.
A short explanation of why you should care...
R has this great feature where you can seamlessly pass arguments between functions. Suppose we have this function that computes the means of the columns in a matrix or data frame:
mean_by_column <- function(x, ...) {
results <- rep(NA, ncol(x))
for(i in 1:ncol(x)) results[i] <- mean(x[, i], ...)
results
}
(aside: don't loop over columns with for. See ?apply, or ?colMeans, instead)
There might be some options to the mean function that we want to pass in but those options might change for different applications. Rather than making different versions of the function for different option combinations, any argument that we pass to mean_by_column that is not one it its arguments (x, in this case) is passed to wherever the three dots appear inside the function. For the function above, they go to mean. Suppose there are missing values in x:
example <- matrix(runif(100), ncol = 5)
example[1, 5] <- NA
example[16, 1] <- NA
mean_by_column(example)
[1] NA 0.4922 0.4704 0.5381 NA
mean has an option called na.rm that will compute the mean on the complete data. Now, we can pass this into mean_by_column even though this is not one of its options.
mean_by_column(example, na.rm = TRUE)
[1] 0.5584 0.4922 0.4704 0.5381 0.4658
Here's why this is relevant. caretSA$fit uses train to fit and possibly tune the model.
function (x, y, lev = NULL, last = FALSE, ...)
train(x, y, ...)
Any options that we pass to safs that are not x, y, iters, differences, or safsControl will be passed to train.
(Even further, any option passed to safs that isn't an option to train gets passed down one more time to the underlying fit function. A good example of this is using importance = TRUE with random forest models. )
So, putting it all together:
set.seed(721)
knn_sa <- safs(x = training[, predVars],
y = training$Class,
iters = 500,
safsControl = ctrl,
## Now we pass options to `train` via "knnSA":
method = "knn",
metric = "ROC",
tuneLength = 20,
preProc = c("center", "scale"),
trControl = trainControl(method = "repeatedcv",
repeats = 2,
classProbs = TRUE,
summaryFunction = twoClassSummary,
allowParallel = FALSE))
To recap:
-
the SA is conducted many times inside of resampling to get an external estimate of performance.
- inside of this external resampling loop, the KNN model is tuned using another, internal resampling procedure.
- the area under the ROC curve is used to guide the search (internally) and to know if the SA has overfit to the features (externally)
- in the code above, when safs is called with other options (e.g. method = "knn"),
- safs passes the method, metric, tuneLength, preProc, and tuneLength, trControl options to caretSA$fit
- caretSA$fit passes these options to train
After external resampling, the optimal number of search iterations is determined and one last SA is run using all of the training data.
Needless to say, this executes a lot of KNN models. When I ran this, I used parallel processing to speed things up using the doMC package.
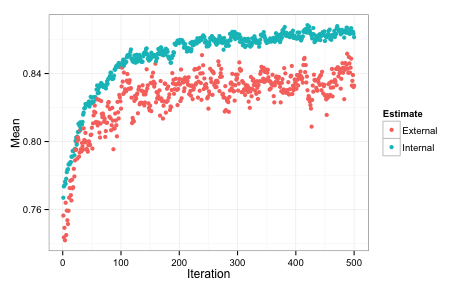
Here are the results of the search:
Simulated Annealing Feature Selection
267 samples
132 predictors
2 classes: 'Impaired', 'Control'
Maximum search iterations: 500
Restart after 25 iterations without improvement (15.6 restarts on average)
Internal performance values: ROC, Sens, Spec
Subset selection driven to maximize internal ROC
External performance values: ROC, Sens, Spec
Best iteration chose by maximizing external ROC
External resampling method: Cross-Validated (10 fold, repeated 5 times)
During resampling:
* the top 5 selected variables (out of a possible 132):
Ab_42 (96%), tau (92%), Cystatin_C (82%), NT_proBNP (82%), VEGF (82%)
* on average, 60 variables were selected (min = 45, max = 75)
In the final search using the entire training set:
* 59 features selected at iteration 488 including:
Alpha_1_Antitrypsin, Alpha_1_Microglobulin, Alpha_2_Macroglobulin, Angiopoietin_2_ANG_2, Apolipoprotein_E ...
* external performance at this iteration is
ROC Sens Spec
0.852 0.198 0.987
The algorithm automatically chose the subset created at iteration 488 of the SA (based on the external ROC) which contained 59 out of 133 predictors.
We can also plot the performance values over iterations using the plot function. By default, this uses the ggplot2 package, so we can add a theme at the end of the call: