I've been looking at this article for a new tree-based method. It uses other classification methods (e.g. LDA) to find a single variable use in the split and builds a tree in that manner. The subtleties of the model are:
- The model does not prune but keeps splitting until achieving purity
- With more than two classes, it treats the data as a two-class system in some parts of the algorithm (but predictions are still based on the original classes)
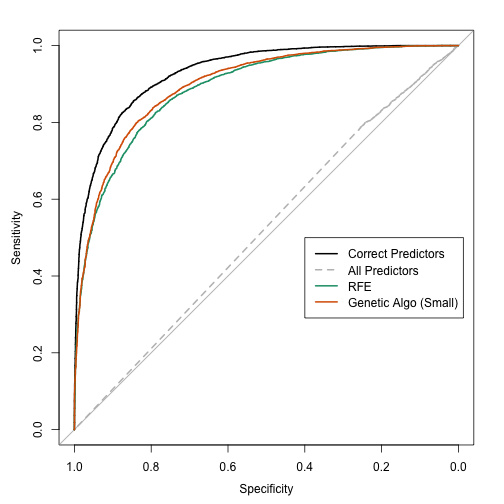
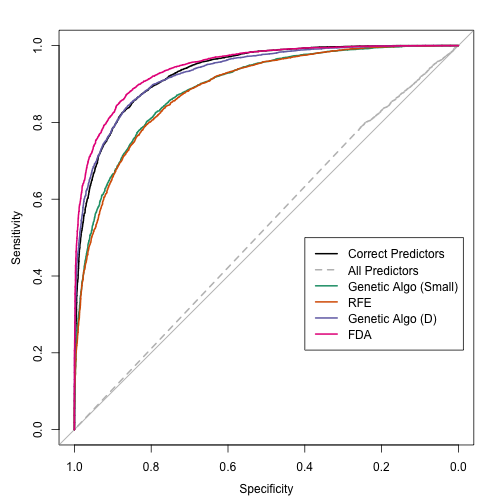
It is similar to oblique trees. These trees look for linear combinations of predictors to use in a split. The similarity between oblique trees and PPtree is the method of finding splits. In each case, a more parametric model can be used for this purpose. Some implementations of oblique trees use PLS, L2 regularization or linear support vector machines to find the optimal combination. Here, the authors use basic discriminant functions but using only a single predictor at a time. This connection wasn't mentioned in the paper (and comparisons were not made to these methods). They compared to CART and random forests. That's disappointing because there are a lot of other tree-based models and we have no idea how this model ranks among them (see Hand's "Classifier Technology and the Illusion of Progress").
My intuition tells me that the PPtree model is somewhat less likely to over-flit the data. While it lacks a pruning algorithm, the nature of the splitting method might make it more robust to small fluctuations in the data. One way to diagnose this is using more comprehensive cross-validation and also assessing whether bagging helps this model. The splitting approach should also reduce the potential problem of bias towards predictors that are more granular. One other consequence of their tree-growing phase is that it eliminates the standard method of generating class probabilities (since it splits until purity).
PPtrees might do a better job when there are a few linear predictors that drive classification accuracy. This could have been demonstrated using simulation of some sort.
A lot of tree methods have sub-algorithms for grouping categorical predictors. This model only works with such data as a set of disconnected dummy variables. This isn't good or bad since I have found a lot of variation in which type of encoding works with different tree methods.
The bad news: the method is available in an R package, but there are big implementation issues (to me at least). The package strikes me as a tool for research only (as opposed to software that would enable PPtrees to be used in practice). For example:
- It ignores basic R conventions (like returning factor data for predictions)
- It also ignores object oriented programming. For example, there is no
predictmethod. That function is namedPP.classify. - Speaking of
PP.classify, you have to trick the code into giving predictions on unknown samples. That is a big red flag to me. - Little things are missing (e.g. no print or plot method). They could have used the partykit package to beautifully visualize the tree.
I've ranted about these issues before and the package violates most of my checklist. Maybe this is just part of someone's dissertation and maybe they didn't know about this list etc. However, most of the items above should have been avoided.